
 Skip to navigation
Skip to navigation
Site Primary Navigation:
- About SDSC
- Services
- Support
- Research & Development
- Education & Training
- News & Events
Search The Site:
| USER RESEARCH | Contents | Next | |
A Third Target in the Fight against AIDS |
|
| FEATURED Senyon Choe, Frederic Bushman, Jason Greenwald,Young Hwang, Vincent Le, Denise Rhodes, Kathleen Rubins, Salk Institute for Biological Studies James M. Briggs, Roberto Lins, Adeymi Adesokan, Thereza A. Soares, University of Houston |
J. Andrew McCammon, Jay Siegel, Heather Carlson, Kevin Masukawa, Tiziana Mordasini, Valentina Molteni, UC San Diego
John Faulkner, Scripps Institution of Oceanography |
|
AN INTEGRASE INHIBITORMOLECULAR DYNAMICSDYNAMIC PHARMACOPHORE MODELINGFINDING POTENTIAL INTEGRASE INHIBITORS |
|
|
|
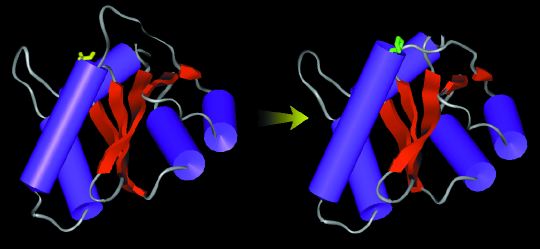 |
|
Molecular dynamics simulations begun with the initial form of the catalytic domain of the HIV-1 integrase (left) ended with the final structure (right), which has a longer, more stable alpha helix (cylinder shape at left in both images). |
|
|
|
AN INTEGRASE INHIBITORThe first drugs for AIDS worked by interfering with the action of the reverse transcriptase enzyme of HIV. They represented a ray of hope, but they had many side effects and seemed to lose efficacy gradually. A breakthrough came several years ago with the advent of drugs that interfered with the action of the HIV enzyme protease. Computer simulation played a key role in the discovery and design of the protease inhibitors. But HIV mutates rapidly, and resistance to all the drugs, including the protease inhibitors, is a growing problem. So scientists are now attacking HIV at a third enzyme, the integrase, which plays an essential role in co-opting the host cell's machinery for viral replication. Computer simulations are proving more important than ever in the search for integrase inhibitors. The effort unites groups from the University of Houston, UC San Diego, Scripps Institution of Oceanography (SIO), and the Salk Institute for Biological Studies in a cooperative, integrated computational and experimental search. The team has found and is testing several potential integrase inhibitors using methods ranging from computational design to organic chemical synthesis. "Computation has been critical in this effort," said computational chemist James M. Briggs, assistant professor in the Department of Biology and Biochemistry at the University of Houston, "both for getting a picture of the structure of the integrase active site and for obtaining information about and testing the binding properties of possible inhibitors." The investigations of six groups at four institutions are funded by a Program Project grant from the National Institutes of Health (NIH), and additional funding for the work of each group comes from both NIH and NSF. Overall leader of the project is Senyon Choe, a crystallographer at the Salk Institute. Other project participants include computational biochemist Briggs and his postdoctoral fellow, Roberto Lins; J. Andrew McCammon, the Joseph Mayer Professor of Chemistry and Biochemistry at UC San Diego, and postdoctoral fellow Heather Carlson in McCammon's group; virologist Frederic Bushman and his group at Salk; marine biochemist John Faulkner and his group at SIO; and Jay Siegel and his group of organic chemical synthesists in the Department of Chemistry and Biochemistry at UC San Diego. At any given time, team members are working on some piece of this problem. Choe and his group are contributing basic crystallographic studies of the structure of the HIV integrase. Briggs and Lins use long molecular dynamics simulations of this structure to refine the picture of the active site to which an inhibitor might bind. Carlson and McCammon have developed a new way of searching for a pharmacophore--the simplest specification of an active compound that might serve as an inhibitor--using the simulations, other high-performance computations, and complex database searching. Bushman, Faulkner, and Siegel test, discover, and synthesize likely inhibitors. |
Top| Contents | Next
REFERENCES Lins, R.D., J.M. Briggs, T.P. Straatsma, H.A. Carlson, J. Greenwald, S. Choe, and J.A. McCammon (1999): Molecular dynamics studies of the HIV-1 integrase catalytic domain, Biophysical Journal 76, 2999-3011. Carlson, H.A., K.M. Masukawa, W.L. Jorgensen, R.D. Lins, J.M. Briggs, and J.A. McCammon (1999): Developing a dynamic pharmacophore model for HIV-1 integrase, Journal of Medical Chemistry (in press). |
|
|
|
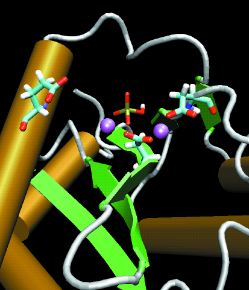 |
Figure 2. Cofactors Bound
The molecular dynamics simulation used two magnesium ions (purple spheres) and a phosphate ion (stick model) in the active site, which is the beginning of an integrase-DNA complex. |
|
|
|
MOLECULAR DYNAMICSIntegrase has been a difficult system for drug development because a complete structure of the enzyme as found in vivo is unavailable. The HIV integrase splices the viral genome into the host cell's DNA, thereby tricking the host cell machinery into making viral proteins. The structure of the entire molecule may be divided into three domains: the N terminus (residues 1-49), which binds zinc; the catalytic core (50-212), which binds magnesium; and the C terminus (213-288), which binds DNA. "Our MD simulations of the catalytic domain were used to complete and refine the crystallographic structure supplied by Senyon Choe," Briggs said. This structure, as well as others that have been published since the studies began, are either incomplete (missing atoms) in the region of the active site or have very high uncertainties in this area, owing to the extraordinary mobility and flexibility of key regions of the structure. Briggs and Lins started the modeling work in 1997 while Briggs was an assistant adjunct professor of pharmacology and Lins a graduate student in the McCammon group at UC San Diego. Their studies were published in the Biophysical Journal this year. Using the SDSC Cray T90 and Cray T3E, they did very long (1-2 nanoseconds) molecular dynamics simulations for the active site without ligands as well as for the site with one or both magnesium ion cofactors bound. Finally, they simulated the catalytic domain with two magnesium ions and a phosphate ion bound at the active site. The ions and phosphate represent a small portion of a DNA backbone that the viral integrase would bind. The process clarified aspects of the active site structure not yet discoverable by crystallography alone (Figures 1-2). "A clear idea of the native structure of HIV integrase at the active site is needed to find or construct suitable inhibitors," Briggs said. |
Top| Contents | Next |
|
|
|
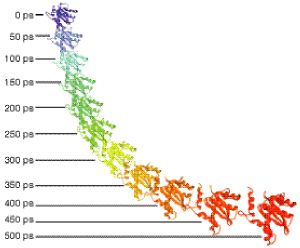 |
Figure 3. Active Site Snapshots
Carlson's simulations used snapshots, taken every 50 picoseconds, from the beginning of the molecular dynamics simulation by Briggs and Lins to bind potential pharmacophore models. Colors correspond to time intervals for probe dockings of Figure 4. |
|
|
|
DYNAMIC PHARMACOPHORE MODELING"Jim's studies were vital to our drug design efforts," said Heather Carlson, who used structures from the simulations as targets for studies to identify the inhibitor pharmacophore, a map of the simplest requirements for a small compound that can be bound in the active site to keep the integrase from binding DNA. For her novel dynamic method, she sampled the active site structure every 50 picoseconds (ps) during the last half of the nanosecond simulations (Figure 3). "The 50-ps snapshots gave us many configurations of the active site to describe some inherent flexibility of the system," Carlson said. She then applied a computational procedure called multi-unit search for interacting conformers (MUSIC), developed during her graduate studies with William Jorgensen at Yale University. MUSIC carries out multiple, simultaneous, gas-phase minimizations for hundreds of probe molecules within the active site. In Carlson's study, methanol molecules were used as probes to define the sites for complementary hydrogen-bond donors that are conserved over the course of the simulation. "The conserved binding sites define the pharmacophore model for an inhibitor," Carlson said, "and the dynamic procedure allows for the same flexible motion seen in the simulation of the unligated core domain containing the active site." (Figure 4). "This is the first use of this method to represent the appropriate flexibility of a protein," McCammon said. "The need for flexibility in ligand docking has been well documented, but the computational challenge means that the large majority of publications continue to report the use of only one static crystal or NMR structure for multiple-copy simulations." |
Top| Contents | Next |
|
|
|
Figure 4. The Dynamic Pharmacophore Models
The overlaid dynamic "dockings" of methanol probes color-coded to correspond to the time snapshot to which they were docked (as in Figure 3), and the time-resolved integrase structures to which they were docked (gray). Each cluster of hydrogen-bond donors contains probes determined using structures from earlier (blue-green) and later (yellow-red) in the simulation. |
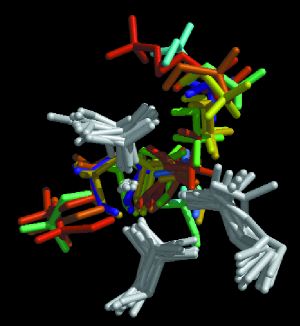 |
|
|
|
FINDING POTENTIAL INTEGRASE INHIBITORSCarlson then screened the large Available Chemicals Database for likely inhibitor compounds using the dynamic model pharmacophores as screening templates. Of 78 compounds found using variants of the dynamic models, small peptides that would already have been found by common high-throughput screening were discarded, as were some compounds too large or reactive to be suitable drugs, leaving 12 candidates. "Our experimental collaborators at Salk, SIO, and UC San Diego are now screening compounds identified from the database searches," Briggs reported. Bushman is testing the candidate inhibitors in his laboratory, using both the compounds found by computation and analogs either found in marine environments by Faulkner or synthesized by Siegel. Choe's group will characterize crystallographically, in complex with the integrase, compounds that Bushman determines are sufficiently active. "We're not there yet, but we're on the track of half a dozen promising integrase inhibitors," Briggs said. "We are finding this complete and concurrent cycle of experimental and computational research to be rapid and effective despite the geographical and institutional separation of our groups. The thought that what has just been discovered in a computation will shortly be tested in an experiment energizes all of us." |
Top| Contents | Next |