PROJECT LEADERS
Charles Brooks
The Scripps Research Institute (TSRI)
Andrew Grimshaw
University of Virginia
PARTICIPANTS
David Case, Michael Crowley
TSRI
Katherine Holcomb
John Karpovich
U. Virginia
Joan Shea
TSRI and University of Chicago
REFERENCE
Shea, J.-E., and C.L. Brooks III. 2000. From folding theories to folding proteins: A review and assessment of protein folding and unfolding calculations. Annual Reviews of Physical Chemistry 52 (in press).
|
Protein Folding in a Distributed Computing Environment
|
 roteins are synthesized within the cells of plants and animals. To function, a newly formed protein must adopt a specific, folded, 3-D shape. This shape, or conformation to biologists, is related to the sequence of amino acids that constitute the protein. The shape is important because it determines how the protein will function--how the protein will bind to other molecules, for example, thus causing or preventing disease. Charles L. Brooks, III, professor of molecular biology from The Scripps Research Institute in La Jolla, and Andrew Grimshaw, professor of computer science from the University of Virginia, lead an NPACI alpha project to distribute large-scale protein-folding problems across high-end resources on a computational grid. roteins are synthesized within the cells of plants and animals. To function, a newly formed protein must adopt a specific, folded, 3-D shape. This shape, or conformation to biologists, is related to the sequence of amino acids that constitute the protein. The shape is important because it determines how the protein will function--how the protein will bind to other molecules, for example, thus causing or preventing disease. Charles L. Brooks, III, professor of molecular biology from The Scripps Research Institute in La Jolla, and Andrew Grimshaw, professor of computer science from the University of Virginia, lead an NPACI alpha project to distribute large-scale protein-folding problems across high-end resources on a computational grid.
|
 |
Can the 3-D structure of a protein be predicted given only its amino acid sequence? What is the pathway from the unfolded to the folded state for any given protein? What is the physical basis for the stability of the folded conformation? These questions constitute the "protein folding problem," and both experimental and computational biologists are trying to find the answers.
"Proteins know how to fold; they do it in a matter of seconds to minutes," Brooks said. But a random search among all possible folding scenarios is an infinite task. Theoretically, folding can be described as the descent of a folding chain down a funnel, with many intermediate configurations at the top, fewer down lower, and the solution--the minimum free energy configuration--at the bottom. The slope of the funnel represents the thermodynamic drive to the native state, while the local roughness of the funnel is the potential for transient trapping of the process in a local energy minimum.
Brooks and his colleagues are using the molecular dynamics programs CHARMM and AMBER to calculate "folding landscapes" for simple proteins. The group performs all-atom molecular dynamics simulations of unfolded proteins in the process of folding. The progress of the calculations may be represented (Figure 1) by a diagram on which one axis represents the molecule compactness (radius of gyration)and the other represents the degree of folding (the fraction of native "contacts"). As the radius of gyration gets smaller and the number of contacts increases, the protein approaches the native, folded state.
|
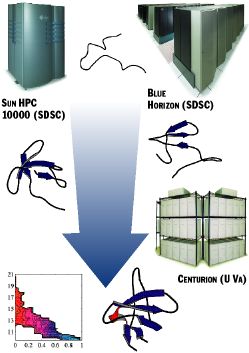
Figure 1. Distributed Protein Folding
The various compute platforms run the CHARMM algorithm independently, starting from distinct intermediate stages of folding (in this case, of a protein domain of the sarcoma virus); a final structure and energy landscape of the folding process are the results.
|
|
|
"Because the most efficient way to do the many calculations required is to distribute them across multiple parallel computing systems, we have been working with the Brooks group to do just that," Grimshaw said. "We have modified CHARMM to work with our metasystem, Legion, across the network, and we've been testing connections between SGI systems at TSRI, Blue Horizon and the Sun HPC 10000 at SDSC, and the Centurion cluster here at Virginia."
One protein system under study is a small protein from the sarcoma virus, composed of five beta-strands (flat, ribbon-like structures), a small helical (spiral) structure, and three simple loops of sequence. "What I found during multiple runs across the distributed system," said Joan Shea of the Brooks group, "was that the folding of the protein is very polarized, with a specific structural region involving a beta hairpin and a turn forming very early on. The formation of this region is key to the folding process."
This result is in line with the idea, shared by Brooks and other biologists, that the kinds of secondary structure present in the folded state (beta sheets, helices, hairpin turns, and loops) determine the overall shape of the folding energy landscape. Additionally, Shea said, they found that water molecules play a key role in "lubricating" the search for the final folded state after the initial structure forms a compact globule. "This is a unique role for water molecules," she said, "and it represents a discovery not yet attainable from experiment." The detailed simulations performed in the project, Brooks noted, are thus leading to an ability to draw conclusions with precision at the level of molecular details.
"Our system is nearly ready for some very ambitious calculations," Brooks said, "and we will soon be inviting other investigators to explore the possibilities of the new technology. The system is growing more robust with each trial, and we anticipate that it can be used not only for the folding problem but also for structural genomics and the assessment of predicted protein structure."
--MM *
|

 Skip to navigation
Skip to navigation